

Introduction
TYK2-rearrangements have recently been detected in high-risk acute lymphoblastic leukemia (HR-ALL) cases and are associated with poor outcome. The resultant fusion protein is predominantly driven by Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling. Thus, JAK/TYK2 inhibitors (JAKi) are among the most promising targeted therapeutics against these fusions. This was confirmed in aMYB-TYK2mouse model where the induced aggressive B-ALL was effectively targeted by the novel dual SYK/JAKi, cerdulatinib (cerd) (Tavakoliet al.2020, EHA abstract EP353). Despite the clinical benefit of JAKi in myeloproliferative neoplasms, resistance occurs resulting in relapse. Hence it is necessary to identify potential JAKi-mediated resistance mechanisms inTYK2-rearranged B-ALL patients. This study modeled cerd resistance mechanisms to recapitulate possible clinical scenarios.
Methods
Ba/F3 pro-B cells were retrovirally transduced with a plasmid construct containing theMYB-TYK2fusion gene isolated from an ALL patient. A cerd resistant line (cerdres) was generated by exposure of Ba/F3-MYB-TYK2cells to increasing concentrations of cerd (up to 3µM; clinically achievable plasma level is 1-2µM) over a period of 151 d. IC50 was determined via CellTiter-Glo proliferation assay. Downstream signaling was determined by phospho-flow analysis. Sanger sequencing was performed over theMYB-TYK2fusion gene to identify emergence of mutations. Site directed mutagenesis of theMYB-TYK2fusion construct was used to model an identified mutationin vitro. Computational modeling of theTYK2mutation and cerd docking was performed via ICM-Pro (Molsoft L.C.C.). The effect of long-term exposure to cerd on activation of JAK family kinases was investigated via western blot. A cerd resensitized line (cerdresen) was generated by culturing cerdres Ba/F3-MYB-TYK2cells in cerd free media for 5 weeks.
Results
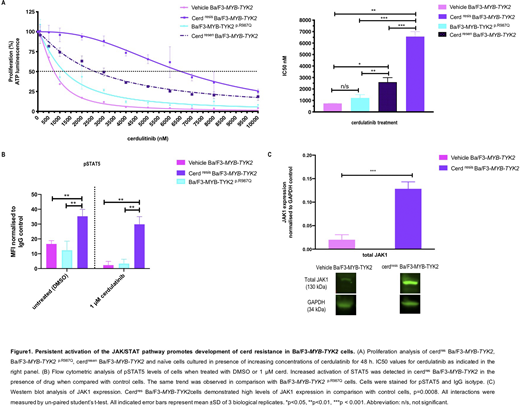
Long-term exposure of Ba/F3-MYB-TYK2cells to cerd resulted in resistance with an 8.7-fold increase in IC50 compared to vehicle control cells (DMSO exposed) (IC50=6508 vs 739nM,p=0.001; Figure 1A). A novel mutation in the kinase domain ofTYK2(p.R987Q, c.3338G>A) was identified in ~50% of cerdres Ba/F3-MYB-TYK2cells. However,de novointroduction ofMYB-TYK2p.R987Q into parental Ba/F3 cells indicated that resistance to cerd was not due to the mutation alone, as these cells displayed no significant decreased sensitivity to cerd compared with control cells (IC50=1200 vs 739nM,p>0.05; Figure A). Computational modeling indicated the binding orientation of cerd to the mutated kinase domain was reversed 180º resulting in less favourable binding (TYK2p.R987Q vsTYK2, binding score= -18.6 vs -23.4). Phosphoflow analysis demonstrated increased JAK/STAT signalling in cerdres Ba/F3-MYB-TYK2compared with control cells (MFI= 35.2 vs 16.5,p=0.008) that persisted despite TYK2 kinase inhibition (MFI= 29.8 vs 2.3,p=0.004; Figure B). Expression of p.R987Q mutation did not result in increased pSTAT5 levels in Ba/F3-MYB-TYK2p.R987Q. Given that JAK2 heterodimerisation with other JAK proteins can lead to JAK/STAT activation and drug persistence (Meyeret al.2017), other kinases may facilitate phosphorylation of TYK2 in cerdres Ba/F3-MYB-TYK2.Western blot analysis confirmed a significant increase in TYK2 phosphorylation (p=0.01) and JAK1 expression (p=0.0008) in cerdres vs control Ba/F3-MYB-TYK2cells (Figure C). Cerd withdrawal resulted in potential resensitization of cerdres Ba/F3-MYB-TYK2to cerd with an associated decrease in IC50 (cerdres vs cerdresen, 6508 vs 2603nM,p=0.0003). However, IC50 levels did not decrease to levels observed in control cells (cerdresen vs control, 2603 vs 739nM, p=0.01), potentially due to increased activation of STAT5 from cerd-induced accumulation of pTYK2 (Tvorogovet al. 2018).
Conclusions
In vitromodeling suggests that persistent JAK/STAT activation is due to changes in TYK2 expression. Possible heterodimer formation with JAK1 in the setting of JAKi -cerd- exposure allows cells to become resistant. Consequently, the novel evidence of resistance mechanisms to JAKi, provide a rationale for the use of other small molecule inhibitors (e.g. HSP90i and HDACi), to potentially retain TYK2 degradation ability in resistant cells. This targeted approach may contribute to the treatment of patient withTYK2-rearranged ALL.
White:Bristol-Myers Squibb:Honoraria, Research Funding;Amgen:Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal